Nonadiabatic H-Atom Scattering Channels on Ge (111) Elucidated by the Hierarchical Equations of Motion #452
Authors
Xiaohan Dan, Zhuoran Long, Tianyin Qiu, Jan Paul Menzel, Qiang Shi, Victor Batista
Abstract
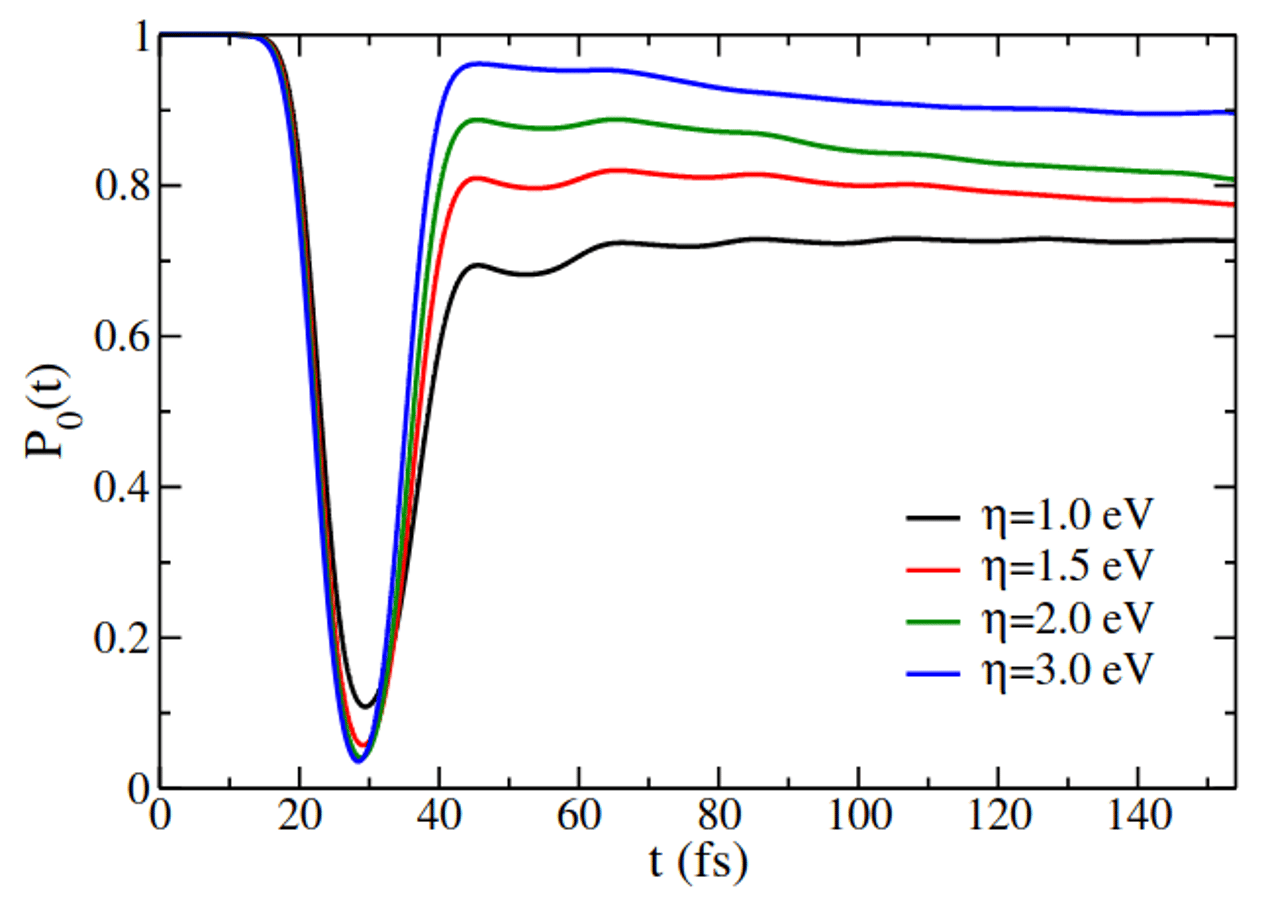
Atomic and molecular scattering at semiconductor interfaces plays a central role in surface chemistry and catalysis, yet predictive simulations remain challenging due to strong nonadiabatic effects causing the breakdown of the Born-Oppenheimer approximation. Here, we present fully quantum simulations of H-atom scattering from the Ge(111)c(2x8) rest site using the hierarchical equations of motion (HEOM) with matrix product states (MPS). The system is modeled by mapping a density functional theory (DFT) potential energy surface onto a Newns-Anderson Hamiltonian. Our simulations reproduce the experimentally observed bimodal kinetic energy distributions, capturing both elastic and energy-loss channels. By systematically examining atom-surface coupling, incident energy, and isotope substitution, we identify the strong-coupling regime required to recover the experimental energy-loss profile. This regime suppresses the elastic peak, implying additional site-specific scattering channels in the observed elastic peak. Deuterium substitution further produces a subtle shift in the energy-loss peak, consistent with experiment. These results establish HEOM as a rigorous framework for quantum surface scattering, capable of capturing nonadiabatic dynamics beyond electronic friction and perturbative approaches.